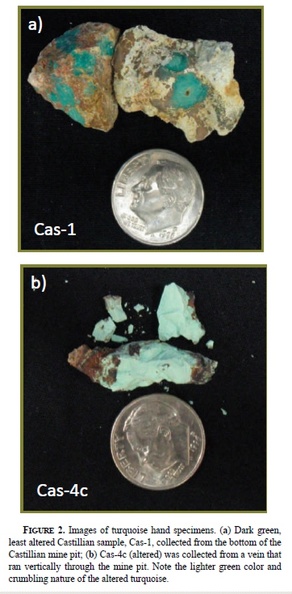
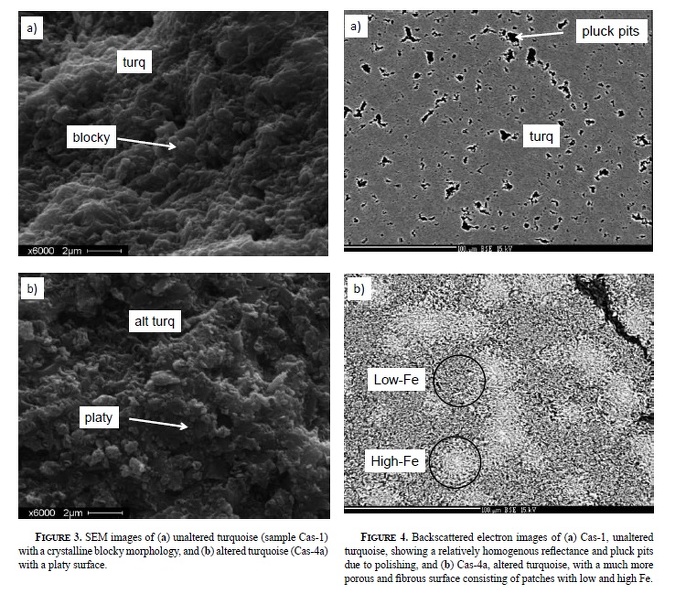
Post by 1dave on Jul 10, 2018 0:20:01 GMT -5
Turquoise = CuAl6(PO4)4(OH)8·4H2O, a hydrated phosphate of copper and aluminum.
Phosphorus is what holds Turquoise together.
This is the structural formula of the phosphoric acid functional group as found in weakly acidic aqueous solution. In more basic aqueous solutions, the group donates the two hydrogen atoms and ionizes as a phosphate group with a negative charge of 3.
Aluminium phosphate Al(PO4) is a natural chemical compound. The anhydrous form is found in nature as the mineral berlinite.

Berlinite looks like quartz and has a structure that is similar to quartz with silicon replaced by Al and P. The AlO4 and PO4 tetrahedra alternate. Like quartz, Al(PO4) exhibits chirality (right and left handedness)and piezoelectric properties. Crystalline Al(PO4) (berlinite) when heated, converts to tridymite and cristobalite forms, and this mirrors the behavior of silicon dioxide.
US Major Copper Mines
The amount of aluminum and copper can vary from all aluminum to 50/50, resulting in the "Planerite-Turquoise Series"
This series is in the middle of the larger series from Al Variscite to Cu Libethenite.

Also other elements such as iron and zinc can enter the structure, replacing aluminum and copper in the A and B positions to various amounts. This increases the Turquoise variability into what is known as the Turquoise group.

Table 1
There we find the Turquoise - Chalcosiderite Series.- Chemical Formula: CuFe6 (PO4)4 (OH)8:4H2O. AKA Variquoise. - Colors: light to dark green.

The crystal structure of turquoise is triclinic, space group P1, and is illustrated in Figure 1.
Different site nomenclatures have been used in many of the structural and spectroscopic studies of the minerals of the turquoise group, depending on the chemical compositions under study.
Here we use a more general site nomenclature that avoids phrases as for example “Fe3+ occupying an Al site”.
There are four sites that are octahedrally coordinated by O2– and (OH)− anions and two T sites that are tetrahedrally coordinated by O2– anions, and are occupied by P. The octahedrally coordinated sites may be divided into two groups, the M1–M3 sites that are occupied dominantly by small trivalent cations, and the X site that is occupied by medium-sized divalent cations (primarily Cu2+, Fe2+, and Zn).
Thus, the structural formula for turquoise is
X (M12M22M32)Σ=6 (PO4)4(OH)8·4H2O.
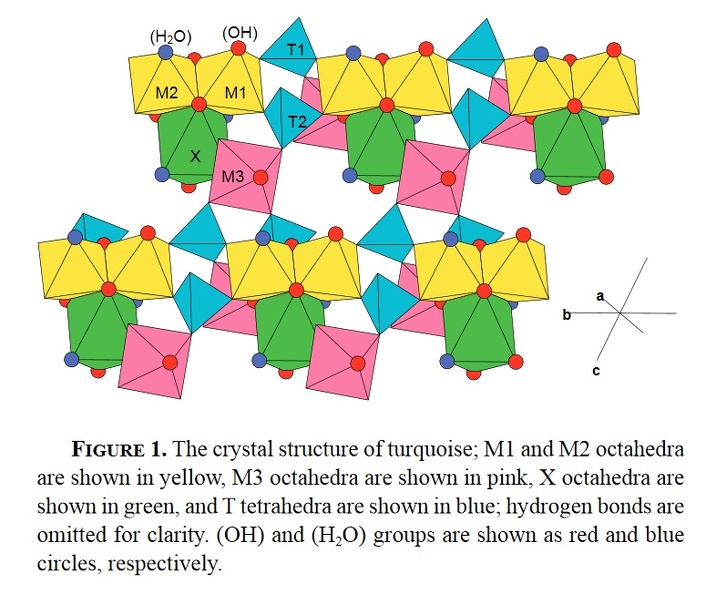
In the structure of turquoise (Fig. 1), pairs of edge-sharing M1 and M2 octahedra are linked by sharing corners with pairs of T tetrahedra to form chains that extend in the b direction. These chains are linked in the a and c directions by sharing corners with M3 octahedra, and further linkage is provided by X octahedra that share edges with the M1 and M2 octahedra. The heteropolyhedral framework is strengthened by a network of hydrogen bonds involving the (OH) groups in the formula. Of particular importance in Figure 1 are the similarities and differences exhibited by the M1–M3 octahedra. The M1 and M2 octahedra are very similar in their general stereochemistry and their linkage to adjacent cation polyhedra is identical. On the other hand, the local stereochemistry of the M3 octahedron is very different from that of M1 and M2. It does not share any edges with the M1 and M2 octahedra and shares a corner with the X octahedron (Fig. 1). Thus similar behavior is expected from constituents occupying the M1 and M2 octahedra and different behavior from constituents occupying the M3 octahedron.
Substitution of Al by Fe3+ results in the solid-solution series turquoise-chalcosiderite: Cu(Al,Fe3+)6(PO4)4(OH)8·4H2O.
Cid-Dresdner and Villarroel (1972) studied rashleighite, an intermediate member of the series, by powder X-ray diffraction, and concluded that Fe3+ is statistically distributed over the three Al sites (i.e., M sites in our notation). However, Giuseppetti et al. (1989) refined the crystal structure of an Al-bearing chalcosiderite (Al = 0.54 apfu) and found that Fe3+ is partly ordered at the Fe1 site (corresponds to Al3 site in the work of Cid-Dresdner 1965). The Al1 and Al2 sites were labeled by Giuseppetti et al. (1989) as Fe2A and Fe2B, respectively. Giuseppetti et al. (1989) also reanalyzed the powder X-ray diffraction data of Cid-Dresdner and Villarroel (1972) for rashleighite and cautiously suggested similar behavior of Fe3+ in intermediate compositions of the series, where it is very difficult to find single crystals suitable for X-ray diffraction.
Phosphorus is what holds Turquoise together.
This is the structural formula of the phosphoric acid functional group as found in weakly acidic aqueous solution. In more basic aqueous solutions, the group donates the two hydrogen atoms and ionizes as a phosphate group with a negative charge of 3.
Aluminium phosphate Al(PO4) is a natural chemical compound. The anhydrous form is found in nature as the mineral berlinite.
Berlinite looks like quartz and has a structure that is similar to quartz with silicon replaced by Al and P. The AlO4 and PO4 tetrahedra alternate. Like quartz, Al(PO4) exhibits chirality (right and left handedness)and piezoelectric properties. Crystalline Al(PO4) (berlinite) when heated, converts to tridymite and cristobalite forms, and this mirrors the behavior of silicon dioxide.
Copper entering the structure is what gives turquoise its special blue color.
US Major Copper Mines
en.wikipedia.org/wiki/Phosphate_mining_in_the_United_States
Geology
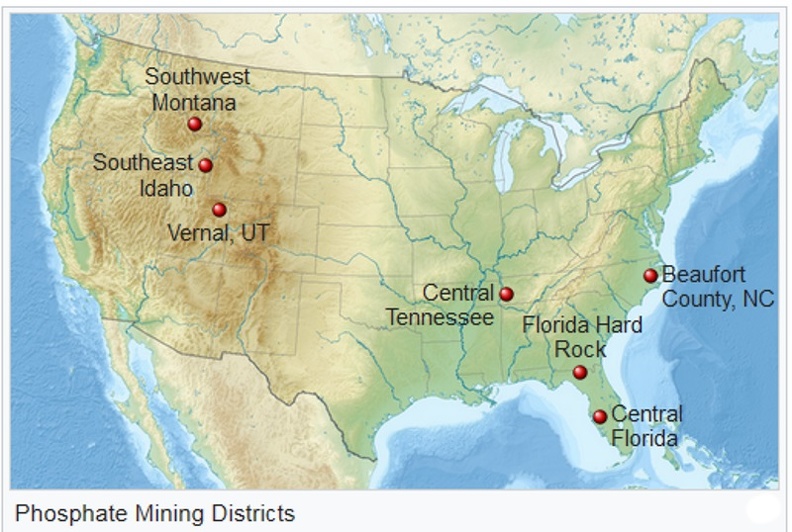
Although some igneous rocks have economic grades of phosphate, no igneous phosphate rock is mined in the US. Small amounts of phosphate were mined from igneous apatite deposits in Virginia, New Jersey, and New York. But since 1948, all phosphate deposits mined in the US have been sedimentary deposits. However, a small amount of phosphate was produced in the 1970s, as a byproduct of iron mining in Missouri.[12]
Most phosphate mining is of sedimentary phosphorite, a phosphorus-rich deposit formed under shallow marine conditions. However, a number of US phosphate deposits occur where the original marine-deposited phosphate minerals have been concentrated by weathering and redeposition, often under nonmarine conditions.
Bone Valley, Florida
The largest source of US phosphate is the Bone Valley, or Central Florida mining district, which produces from the Pliocene-age Bone Valley Formation.[13]
Florida Hard Rock
The Florida Hard Rock district mines phosphate from the Alachua Formation of Pliocene age. The Alachua Formation is nonmarine and consists of sediments rich in phosphate that was weathered from the Hawthorne Formation of Miocene age.[13]
North Carolina
The North Carolina phosphate deposit occurs in the Pungo River Formation of Miocene age.<[14] The phosphate was deposited under shallow marine conditions.[15]
South Carolina
The South Carolina river-pebble deposits formed along modern rivers which reworked and concentrated lower-grade phosphate deposits.
Tennessee
Three classes of phosphate deposits have been mined in central Tennessee; in order of decreasing importance: brown phosphate rock, blue phosphate rock, and white phosphate rock.[16]
The most important phosphate deposits were the so-called brown phosphate rock, mined from Ordovician limestones, some parts of which are rich in phosphate. The highest-grade deposits were found where weathering or shallow groundwater had preferentially dissolved away the calcium carbonate. The last mine in Tennessee, which produced phosphate rock from the Bigby-Cannon formation, closed in 1991.
The blue phosphate rock was found in the Devonian-age Chattanooga Shale.
Minor phosphate production was mined from white phosphate rock, which formed in karsted Silurian limestones.
Western US, Phosphoria
The western deposits, in Utah, Wyoming, Idaho, and Montana, are in the Phosphoria Formation of Permian age. The phosphate deposits occur in two members of the Phosphoria: the Retort phosphatic shale member in the upper part of the formation, and the Meade Peak phosphatic shale member in the lower part of the formation.
Northeast Utah Mississippian
Although not presently mined, phosphate deposits occur in the Brazer limestone of Mississippian age in Utah.
Geology
Although some igneous rocks have economic grades of phosphate, no igneous phosphate rock is mined in the US. Small amounts of phosphate were mined from igneous apatite deposits in Virginia, New Jersey, and New York. But since 1948, all phosphate deposits mined in the US have been sedimentary deposits. However, a small amount of phosphate was produced in the 1970s, as a byproduct of iron mining in Missouri.[12]
Most phosphate mining is of sedimentary phosphorite, a phosphorus-rich deposit formed under shallow marine conditions. However, a number of US phosphate deposits occur where the original marine-deposited phosphate minerals have been concentrated by weathering and redeposition, often under nonmarine conditions.
Bone Valley, Florida
The largest source of US phosphate is the Bone Valley, or Central Florida mining district, which produces from the Pliocene-age Bone Valley Formation.[13]
Florida Hard Rock
The Florida Hard Rock district mines phosphate from the Alachua Formation of Pliocene age. The Alachua Formation is nonmarine and consists of sediments rich in phosphate that was weathered from the Hawthorne Formation of Miocene age.[13]
North Carolina
The North Carolina phosphate deposit occurs in the Pungo River Formation of Miocene age.<[14] The phosphate was deposited under shallow marine conditions.[15]
South Carolina
The South Carolina river-pebble deposits formed along modern rivers which reworked and concentrated lower-grade phosphate deposits.
Tennessee
Three classes of phosphate deposits have been mined in central Tennessee; in order of decreasing importance: brown phosphate rock, blue phosphate rock, and white phosphate rock.[16]
The most important phosphate deposits were the so-called brown phosphate rock, mined from Ordovician limestones, some parts of which are rich in phosphate. The highest-grade deposits were found where weathering or shallow groundwater had preferentially dissolved away the calcium carbonate. The last mine in Tennessee, which produced phosphate rock from the Bigby-Cannon formation, closed in 1991.
The blue phosphate rock was found in the Devonian-age Chattanooga Shale.
Minor phosphate production was mined from white phosphate rock, which formed in karsted Silurian limestones.
Western US, Phosphoria
The western deposits, in Utah, Wyoming, Idaho, and Montana, are in the Phosphoria Formation of Permian age. The phosphate deposits occur in two members of the Phosphoria: the Retort phosphatic shale member in the upper part of the formation, and the Meade Peak phosphatic shale member in the lower part of the formation.
Northeast Utah Mississippian
Although not presently mined, phosphate deposits occur in the Brazer limestone of Mississippian age in Utah.
The amount of aluminum and copper can vary from all aluminum to 50/50, resulting in the "Planerite-Turquoise Series"
Planerite Al6(PO4)2(PO3OH)2(OH)8•4H2O - Turquoise CuAl6(PO4)4(OH)8·4H2O
With Planerite varying in Color from White, verdigris-green, lime-green, pale blue-green to olive-green.
This series is in the middle of the larger series from Al Variscite to Cu Libethenite.
Also other elements such as iron and zinc can enter the structure, replacing aluminum and copper in the A and B positions to various amounts. This increases the Turquoise variability into what is known as the Turquoise group.
AO-1B6(P04)4_xCP030HMOH)s-4H20, where x varies from 0-2
or A0–1B6(PO4)4(OH)8⋅4H2O
Table 1
with Cu2+ or Fe2+ as the most common constituents at the A position and Al3+ and Fe3+ at the B position. However, Ca2+ or Zn2+ can occur at the A position in some of the more rare members of the turquoise group. The range in chemical composition (i.e., different concentrations of Cu, Fe, and Al) of turquoise (sensu lato) produces a wide range of colors (Table 1). Foord and Taggart (1998) suggest that differences in the Cu and Fe ratio are responsible for the differences in color of the samples: blue turquoise has Cu at the A position and Al at the B position, whereas green turquoise (chalcosiderite)largely contains Fe3+ at the B position.
There we find the Turquoise - Chalcosiderite Series.- Chemical Formula: CuFe6 (PO4)4 (OH)8:4H2O. AKA Variquoise. - Colors: light to dark green.
The crystal structure of turquoise is triclinic, space group P1, and is illustrated in Figure 1.
Different site nomenclatures have been used in many of the structural and spectroscopic studies of the minerals of the turquoise group, depending on the chemical compositions under study.
Here we use a more general site nomenclature that avoids phrases as for example “Fe3+ occupying an Al site”.
There are four sites that are octahedrally coordinated by O2– and (OH)− anions and two T sites that are tetrahedrally coordinated by O2– anions, and are occupied by P. The octahedrally coordinated sites may be divided into two groups, the M1–M3 sites that are occupied dominantly by small trivalent cations, and the X site that is occupied by medium-sized divalent cations (primarily Cu2+, Fe2+, and Zn).
Thus, the structural formula for turquoise is
X (M12M22M32)Σ=6 (PO4)4(OH)8·4H2O.
In the structure of turquoise (Fig. 1), pairs of edge-sharing M1 and M2 octahedra are linked by sharing corners with pairs of T tetrahedra to form chains that extend in the b direction. These chains are linked in the a and c directions by sharing corners with M3 octahedra, and further linkage is provided by X octahedra that share edges with the M1 and M2 octahedra. The heteropolyhedral framework is strengthened by a network of hydrogen bonds involving the (OH) groups in the formula. Of particular importance in Figure 1 are the similarities and differences exhibited by the M1–M3 octahedra. The M1 and M2 octahedra are very similar in their general stereochemistry and their linkage to adjacent cation polyhedra is identical. On the other hand, the local stereochemistry of the M3 octahedron is very different from that of M1 and M2. It does not share any edges with the M1 and M2 octahedra and shares a corner with the X octahedron (Fig. 1). Thus similar behavior is expected from constituents occupying the M1 and M2 octahedra and different behavior from constituents occupying the M3 octahedron.
Substitution of Al by Fe3+ results in the solid-solution series turquoise-chalcosiderite: Cu(Al,Fe3+)6(PO4)4(OH)8·4H2O.
Cid-Dresdner and Villarroel (1972) studied rashleighite, an intermediate member of the series, by powder X-ray diffraction, and concluded that Fe3+ is statistically distributed over the three Al sites (i.e., M sites in our notation). However, Giuseppetti et al. (1989) refined the crystal structure of an Al-bearing chalcosiderite (Al = 0.54 apfu) and found that Fe3+ is partly ordered at the Fe1 site (corresponds to Al3 site in the work of Cid-Dresdner 1965). The Al1 and Al2 sites were labeled by Giuseppetti et al. (1989) as Fe2A and Fe2B, respectively. Giuseppetti et al. (1989) also reanalyzed the powder X-ray diffraction data of Cid-Dresdner and Villarroel (1972) for rashleighite and cautiously suggested similar behavior of Fe3+ in intermediate compositions of the series, where it is very difficult to find single crystals suitable for X-ray diffraction.